ChAT
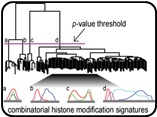
ChAT - unsupervised algorithm to search combinatorial chromatin signatures.
Downloads
Scripts
- ChAT_package.tar.gz - ChAT Scripts
Readme
A. Preparation:
In order to run ChAT, you need to:
- Download the compressed folder ChAT_package.tar.gz
- Decompress the folder. There are three files within the created folder: A) ChAT, B) clustering.R and C) cluster_figure.R.
- Make sure these files are always kept in the same folder.
- Make sure R program is already installed on your computer.
- Check the shebang line of the ChAT file and correct it by the path of env of your computer.
- Add the directory of the folder ChAT_package into the PATH.
B. Running ChAT:
The command line for ChAT is:
$ ChAT -i [the directory where the set of input files located] -o [the output directory where both the final and intermediate files located] -m [the file of the list of histone modifications] -d [the file of the list of critical histone modifications used for initial grouping] -c [the file of the list of chromosomes] -p [p-value threshold to cut the hierarchical tree] -b [bin size]
The detailed explanations of the parameters can be found in Section 4.
One example of running ChAT is:
$ ChAT -i /home/CD4_sample_data -o sample_pattern -m mark_name.txt -d critical_mark.txt -c chromosome.txt -p 0.05 -b 200
This command takes the Wiggle format histone modification files (must be named as *.wig) located in /home/CD4_sample_data folder as the inputs and create the directory sample_pattern to store all the final and intermediate results. mark_name.txt contains the list of histone modifications (each row has a histone modification name) that are consistent with the file names in the input directory. critical_mark.txt contains a subset of histone modifications used for initial grouping. chromosome.txt contains a list of chromosomes (each row has a chromosome name that are consistent with the chromosome names in the wiggle format input files) under consideration. The p-value threshold used to cut the hierarchical tree is set as 0.05. The bin size is set as 200bp using -b.
C. File Format:
(A) Input files
Corresponding to each individual histone modification, there is a Wiggle format file of the ChIP-seq data. All of the files need to be named as histone_mark_name.wig. For example, H3K36me3.wig for H3K36me3. All the files must be stored in the same directory. The name of the directory is the most important parameter for ChAT.
A file of the list of all the histone modifications under consideration need to be provided. Each row has the name of a histone modification.
A file of the list of critical histone modifications for initial grouping need to be provided. Those modifications are important marks based on a priori biological knowledge. This list must be a subset of the modifications under consideration. Each row has the name of a critical histone modification.
A file of the list of all the chromosomes under consideration need to be provided. Each row has the name of a chromosome. The names need to be consistent with the chromosome names in the input wiggle format files.
(B) Output files
All the output files are stored in the created folder specified by -o. The most important final results are saved in 2 folders.
The BED format tracks of genomic locations sharing specific combinatorial chromatin signatures are stored in BED_tracks.
The average histone modification profiles of each signature and the corresponding enrichment curves in PDF files are stored in Signature_info.
D. Parameters:
-i: The directory where all the wiggle format input files (one file for each histone modification) are located. The wiggle format files must be names as *.wig.
-o: The output directory where all the final and intermediate results are stored.
-m: The file with the list of histone modifications under consideration. Each row has the name of one histone modification. They need to be consistent with the name of the wiggle format input files.
-d: The file with the list of critical histone modifications used for initial grouping. Each row has the name of one histone modification.
-c: The file with the list of chromosomes under consideration. Each row has the name of one chromosome. They need to be consistent with the chromosome names in the wiggle format input files.
-p: The p-value threshold used to cut the hierarchical tree, default value: 0.05.
-b: The size of bin, default value 200 (bp).
-h,-help: Display brief explanations of parameters.